
If you’re planning to launch a medical device in the United States, FDA 510(k) clearance is one of the first regulatory hurdles you’ll encounter. But what exactly is 510(k) clearance? How does it differ from FDA approval and certification? And how can manufacturers navigate the process efficiently to enter the US market?
What is FDA 510(k) Clearance?
The FDA 510(k) process, also known as Premarket Notification, is a requirement for most Class II medical devices and some Class I devices that are not exempt. It demonstrates that your new device is substantially equivalent to an existing legally marketed device, called a predicate device.
Once the FDA determines substantial equivalence, your product is “cleared” for commercial distribution in the US.
Important Note: 510(k) clearance is not the same as FDA approval. Approval generally refers to PMA (Premarket Approval) for high-risk Class III devices.
Companies seeking 510(k) clearance aim to prove their device’s safety and effectiveness, often termed as “substantial equivalence.” A device is substantially equivalent if it shares the same intended use and technological characteristics as a predicate device.
After getting 510(k) clearance, the company can start selling the device right away. However, they should be ready for an FDA inspection of their manufacturing processes (called a quality system inspection) at any point after getting the clearance.
Who Needs a 510(k) Submission?
You need to submit a 510(k) if:
- You’re introducing a new Class II device to the U.S. market.
- You’re making significant changes to an existing device that could impact its safety or effectiveness.
- You’re relabeling or rebranding a device for U.S. distribution.
Some Class I devices are exempt, but it’s essential to consult a regulatory expert to determine your specific obligations.
Want to Know More About FDA 510k Clearance?
What’s Inside a 510(k) Submission?
An FDA 510(k) submission typically includes:
- Device description and intended use
- Predicate device comparison (substantial equivalence)
- Performance data, including bench, animal, or clinical testing (if applicable)
- Labeling and instructions for use
- Risk analysis
- Sterilization and biocompatibility information
- Software validation (if applicable)
FDA 510(k) Review Process: Step-by-Step
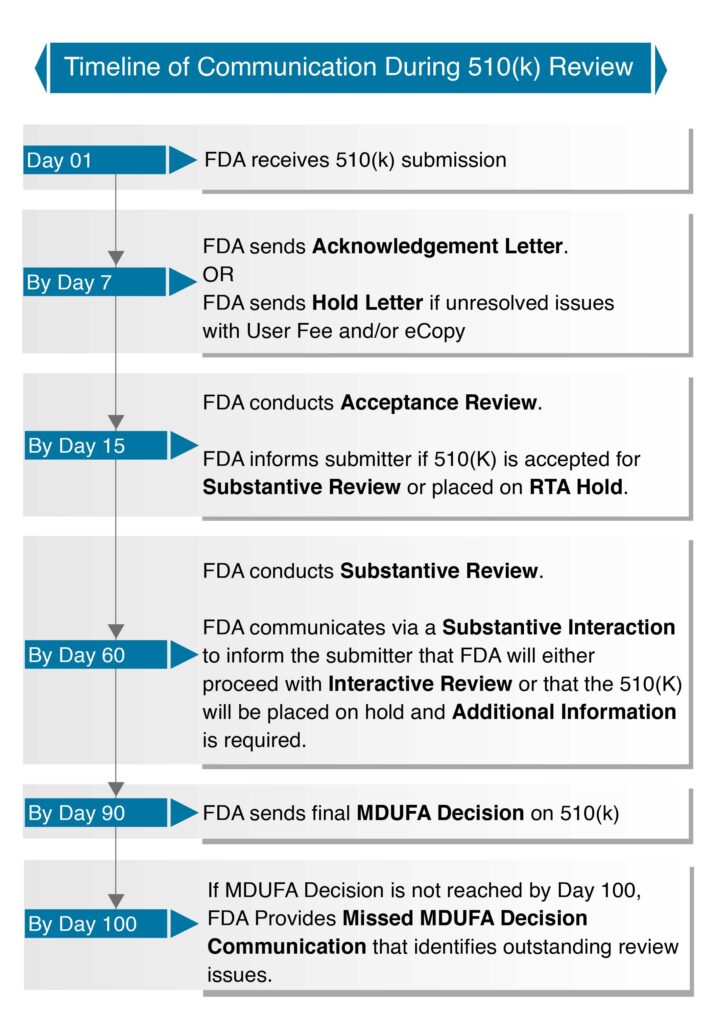
The FDA created a chart as part of MDUFA III to meet 510(k) performance goals. This chart outlines a 90-day timeframe for most clearance decisions and explains communication expectations with FDA reviewers during the medical device registration process in the U.S.
However, it doesn’t precisely mirror the FDA’s review history. Nonetheless, it does offer applicants a clearer idea of what to anticipate from the U.S. regulator as they initiate their registrations.
Common Misconceptions: Clearance vs. Approval vs. Certification
| Term | Meaning | Applies To |
|---|---|---|
| 510(k) Clearance | FDA determines your device is substantially equivalent to a predicate device | Class II (and some Class I) |
| FDA Approval | FDA reviews detailed clinical evidence for safety and effectiveness | Class III (PMA pathway) |
| Certification | Refers to quality standards like ISO 13485, not an FDA process | Global QMS, not part of FDA 510(k) clearance |
How Much Does it Cost to Get 510k Approval?
The cost of getting US FDA 510 (k) depends on the type of submission and the size of your business. According to the FDA, the standard fee for 510 (k) submissions is $19,870 for FY2023, but it can be reduced to $4,967 if you qualify as a small business with gross receipts of sales less than $100 million USD.
If you’re seeking a complete breakdown of the user fee costing structure, this source is your go-to.
How Operon Strategist Helps with FDA 510(k) Submissions
At Operon Strategist, we simplify the complexities of the FDA 510(k) process with our end-to-end regulatory consulting services. Whether you’re a startup or an established manufacturer, we help you:
- Determine product classification
- Identify predicate devices
- Compile and review your submission dossier
- Respond to FDA queries
- Build a compliant quality management system (QMS)
- Prepare labeling and IFUs
Our team of experts has helped manufacturers across the globe successfully achieve FDA 510(k) clearance—quickly and efficiently.
To discuss your support need you can contact us, enquiry@operonstrategist.com or you can WhatsApp us on 8767980322 your queries we answer them shortly.

