US FDA 21 CFR 820.30 Design Control Requirements
Ensure FDA compliance with expert-guided design control systems tailored for your medical device.
At Operon Strategist, we help medical device manufacturers in the USA navigate the critical FDA requirement under 21 CFR 820.30 – Design Control. Whether you’re launching a new product or improving an existing one, our consulting ensures your design and development process is fully compliant, audit-ready, and aligned with your time-to-market goals.
👉 Contact Us to ensure your Design Control process meets FDA expectations.
What is US FDA 21 CFR 820.30 Design Control?
21 CFR 820.30 is a section of the US FDA’s Quality System Regulation (QSR) that outlines the requirements for medical device design and development. It applies primarily to Class II and III devices and focuses on:
- Documented planning and control of design activities
- Clear design inputs and traceable outputs
- Verification, validation, and design reviews
- Risk management and usability
- Maintaining a complete Design History File (DHF)
Failure to comply can lead to FDA 483 observations, warning letters, or product recalls. That’s where we come in.

Design Control Applies To:
- Class II & Class III medical devices
- Certain Class I devices (with design control requirements)
- Drug-device combination products (21 CFR Part 4)
Design Control 21 CFR 820.30 Process for Medical Devices:
We help you implement each phase of the design control process:
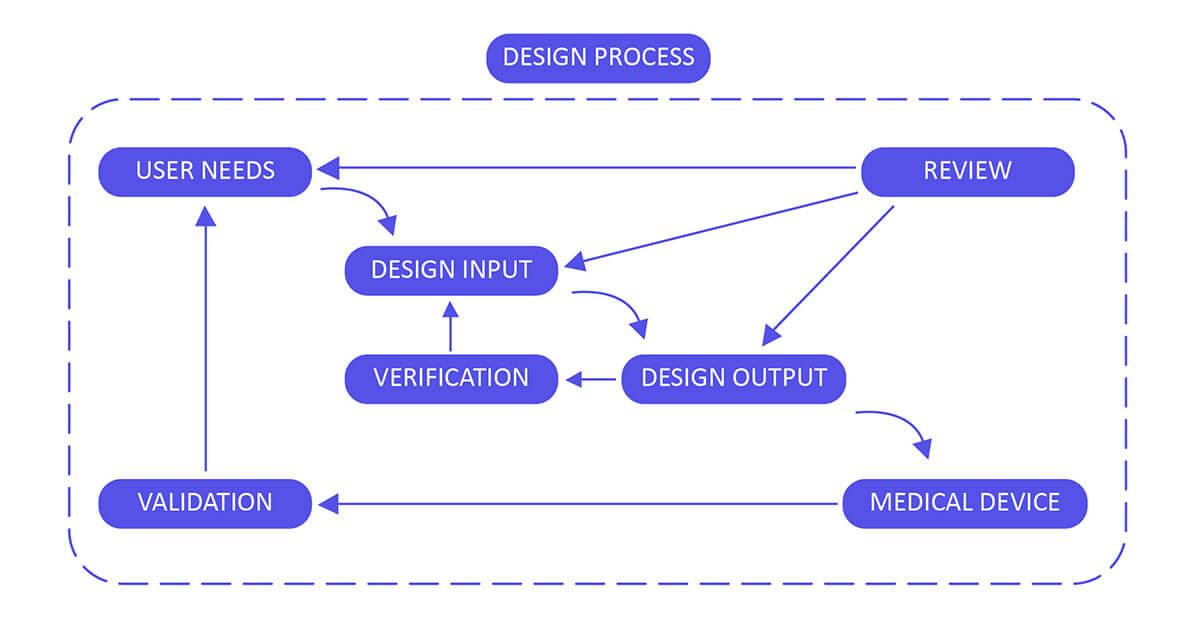
Design Input
Utilize performance, safety, business economics, outputs of risk management, and regulatory requirements as a basis to plan the device with the goal that its motivation and the proposed use are clear. The input may also come from surveying your customers(for example, clinicians, nurses, and patients).
Design Output
Design output methods or particulars need to stipulate or refer to the design input document developed by the team and need to identify the critical measures/outputs for the best possible capacity of the device. These incorporate the tests and strategies that may have been produced, adjusted or used to show conformance with the characterized configuration inputs. Examples of design outputs may include:
- The device itself.
- The user manual.
- Specifications A Risk Analysis Study results (For examples, validation and biocompatibility studies, storage).
- Technical Files.
Design Review
Confirm the design, or identify at an opportune time and right any insufficiencies distinguished at other plan and improvement phases. Two common types of review are hazard analysis, and failure mode and effect analysis.
Design Verification
Confirm the device outline by means of examination and target prove, verify that the design outputs meet the plan inputs. Design verification activities must be arranged and routinely analyzed and the outcomes must be documented.
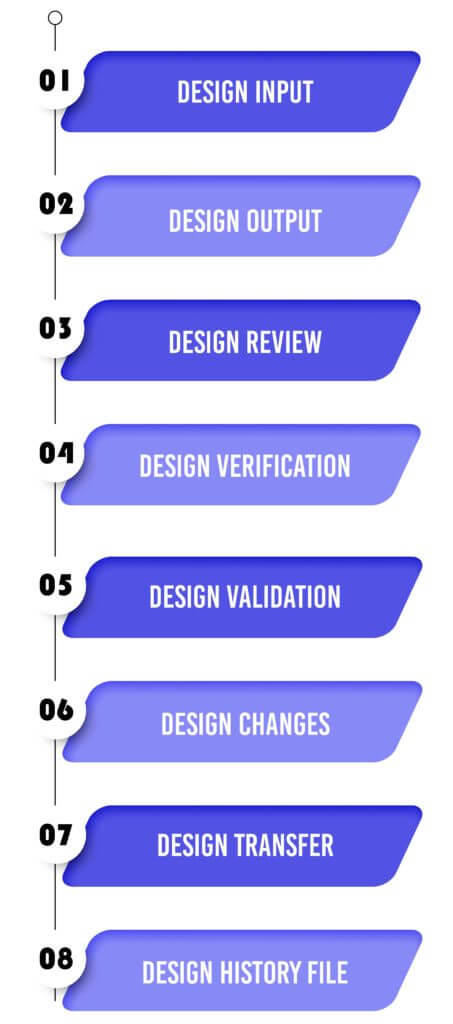
Design Validation
Validate the device design through examination and targeted testing to ensure that the final design output consistently meets the specified intended use. Design validation should follow successful design verification. While design verification is conducted during the design process, design validation ensures that the medical device fulfills its intended purpose. Typically, this is achieved through in vitro performance, functional testing, animal testing, and/or in vivo clinical evaluations and trials.
Design Changes
Guarantee that all plan changes are distinguished, documented, approved, verified, reviewed and endorsed before usage.
Design Transfer
Ensure that the design of the medical device can be correctly translated into production specifications (that is, advancing successfully from product development to manufacturing).
Design History File
The design history file (DHF) aggregates confirm (that is, the history of the design) that demonstrates that the outline was created as per the outline of design control― specifically, the design and development plan, or the outline change design.
Why Choose Operon Strategist for 21 CFR 820.30 Compliance?
We bring deep regulatory expertise and practical consulting to every project. Whether you’re launching a new product or enhancing your existing QMS, we provide tailored solutions to meet your business and compliance goals.
With Operon Strategist, You Get:
- 15+ years of experience in FDA regulatory consulting
- Specialists in 21 CFR 820, ISO 13485, and CE marking
- Served 250+ clients globally, including US-based startups and manufacturers
- Turnkey solutions that save time, money, and reduce regulatory risk
We also support Drug-Device Combination Products under 21 CFR Part 820 and 21 CFR Part 4.
Ready to Streamline Your Design Control Process?
Whether you’re creating a new device or upgrading your QMS for FDA compliance, Operon Strategist is your trusted partner.
👉 Contact Us Today
FAQs
Are Design Controls Part of QMS?
Design Controls is one process that should be integrated into the quality management system (QMS). Your QMS should also have procedures in place to manage documents, records, and suppliers, so Design Controls link those pieces together with additional development specific processes.
What is ISO 13485 Vs 21 CFR 820?
ISO: ISO 13485 is the worldwide acknowledged standard by the International Standards Organization for medical device Quality Management System. The standard states the things required for QMS that helps companies execute and exhibit the abilities to provide high-quality medical devices that reach up to customers and regulatory requirements. ISO 13485 can be used by any company involved in the transfer of medical devices, providing, support phases. Furthermore, it can also be used by external or internal auditors to support the inclusive audit process.
21 CFR Part 820: FDA 21 CFR Part 820: covers up the process used in & the facilities and controls used for the design, manufacture, packaging, labelling, storage, installation, & servicing of medical devices. Manufacturers are inspected by the US FDA as per Part 820 & only compliance with the requirements is assessed. FDA 21 CFR known as the Quality System Regulation QSR acknowledges Current Good Manufacturing Practices CGMP regulations that govern the methods used in and the services, and controls used for, the design, manufacturing, packaging, labelling, storage, installation and servicing for all finished devices willful for human use. These requirements are necessary to ensure that medical devices are safe and effective for use. Medical device manufacturers undergo FDA inspections to assure FDA 21 CFR 820 agreement.